

SBDD Bits
Brief description of recent literature on Structure-Based Drug Design (SBDD)
by Elisa Martino
Crystal Structure Based Mutagenesis of Cattleyene Synthase Leads to the Generation of Rearranged Polycyclic Diterpenes
In their latest paper, Xing, Xu et al. used a structure-based approach to engineering the enzyme Cattleyene Synthase (CyS), which mediates the synthesis of some diterpenes. Diterpenes are chemical scaffolds often used in drug discovery, so more efficient and green synthetic routes for their production are highly desirable. CyS belongs to the family of type I diterpene synthases, a class of enzymes for which just a few structures have been solved.
The authors reported the structure of CyS from Streptomyces cattleya in a complex with its native substrate geranylgeranyl pyrophosphate (GGPP). They identified the interactions responsible for the binding mode of the substrate inside the active site, and they used this structural information to model the poses of reaction intermediates inside the CyS pocket to clarify the catalysis mechanism of the enzyme. Then, they rationally modified specific residues inside the active site, with the aim of elucidating their role in catalysis. Interestingly, some of these mutants did not lead to the production of the natural product, but to a set of different, unprecedented diterpenes.
To understand these results, they produced the crystal structure of the C59A variant of the enzyme, which elucidated the structural reasons for the observed expanded scaffold diversity: in the wild-type CyS, the cysteine (C59) locks a phenyl ring (F86) inside the active site, hindering the pocket, while with the C59A mutation this interaction is disrupted and the phenyl moves away, widening the active site cavity and allowing the production of bigger chemical scaffolds.

Conformational-Design-Driven Discovery of EZM0414: A Selective, Potent SETD2 Inhibitor for Clinical Studies
In their recent paper, Epizyme reported the discovery of a first-in-class, oral inhibitor of the epigenetic target SETD2. With a structure-based, conformational-design-driven modality they developed EZM0414, which is now under Phase 1 study for the treatment of relapsed or refractory (R/R) multiple myeloma (MM) and R/R diffuse large B-cell lymphoma (DLBCL).
SETD2 (SET domain-containing protein 2) is a histone methyltransferase and a potential target for some types of cancer. They previously reported the x-ray crystal structure of SETD2 in a complex with one of the hits compounds from their HTS. Even if supported by structural data from the very beginning of their optimization, they struggled to obtain compounds that translated the observed good in vitro activity into in vivo. They attributed this poor behavior to the lipophilicity imparted by the aromatic ring in the tested molecules, so they replaced it with a saturated system.
In this last paper, they illustrate the conformational studies that led to the identification of the strongly preferred stereoisomer (1R,3S). From the comparison of the protein in complex with the cis and trans isomers, they confirmed their prediction that the cis stereoisomer better occupies the binding pocket, mimicking the binding mode of the planar aromatic ring. The selected stereoisomer showed good activity and physiochemical properties both in vitro and in vivo.
This work shows once again that chiral and saturated compounds are often more successful in clinical translation. This is associated with better solubility profiles and their specificity in the binding mode with the target.

Research & Image Source: https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00167?ref=PDF
Design and Discovery of MRTX0902, a Potent, Selective, Brain-Penetrant, and Orally Bioavailable Inhibitor of the SOS1:KRAS Protein–Protein Interaction
In their latest paper, Mirati Therapeutics reported the design of a disruptor of SOS1:KRAS protein-protein interaction (PPI) that showed promising antitumoral activity, in combination with their covalent KRAS G12C inhibitor (adagrasib). This discovery opens new therapeutic opportunities for the treatment of KRAS-mutated tumors, which are among the most aggressive types (non-small-cell lung, colorectal, and pancreatic).
SOS1 is a guanidine nucleotide exchange factor that mediates the “on-off” switch of GTPase KRAS, favoring its active, GTP-bound state. Since pathological effects of KRAS are associated with its active state, interfering with the formation of the SOS1:KRAS complex to inhibit KRAS activation is an attractive therapeutic strategy. PPI are usually difficult to target with small molecules, but the catalytic site of SOS1 is located in the proximity of the SOS1:KRAS interface, offering a druggable pocket for starting the design of a PPI inhibitor.
With a structure-based approach, the team firstly identified and validated a new scaffold for SOS1 binding: a phthalazine ring, which forms an unprecedently observed salt bridge with E902 in the pocket. Moreover, they investigated several moieties to modify the C-7 substituent, which directly protrudes into the interface between KRAS and SOS1, in the attempt to disrupt the PPI involved in the formation of the complex (R73 on KRAS; N879 and Y884 on SOS1). Finally, structure-activity relationship (SAR) analyses were also performed on positions C-4, C-1, and C-6, to improve potency, metabolic persistency, and bioavailability. The optimization was supported by X-ray co-crystal structures, which guided the rational design of a final molecule with 300-fold increased potency, brain penetrant, and orally bioavailable.
https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c00741?ref=PDF

Research & Image Source: https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c00741?ref=PDF
A Small-Molecule Oral Agonist of the Human Glucagon-like Peptide-1 Receptor
In their recent publication, Griffith et al. from Pfizer undisclosed the drug discovery process that led to the identification of danuglipron, a small-molecule agonist of glucagon-like peptide-1 receptor (GLP-1 R) currently under clinical trial (Phase 2) for the treatment of diabetes mellitus-type 2 and obesity. This is the first report of effective glucose decrease accomplished with a non-peptidic, orally-available agonist of GLP-1 R.
A key step leading to the discovery of the molecule was the set-up of a sensitized high-throughput screening (HTS). This allowed them to identify hit structures that would have been classified as inactive with traditional HTS methods, having very little affinity even to the sensitized-GLP-1 R. With a ligand-driven optimization, they identified the final molecule which showed good solubility, potency, and selectivity.
When conducting tests for selecting animal models, they noticed that the drug was active on primate cells but not on rodents. From sequence comparison and in vitro testing, they identified a species-specific residue, W33, which seemed crucial for the drug activity. To clarify its role, they determined a cryo-EM structure of the complex, which showed how the W33 is necessary for binding the drug since it closes the upper part of the binding pocket. This is a nice example of how structural studies can support drug discovery in the elucidation of the mechanism of action, in this case, the observed species-selectivity.

Research & Image Source: https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c01856
Read more about the danuglipron’s mechanism of action in our Protein of the Month on GLP1-R
Structural basis for SHOC2 modulation of RAS signalling
Another example of the amazing contribution of cryo-EM to our molecular understanding of cellular pathways and to the identification of new therapeutic targets: https://www.nature.com/articles/s41586-022-04838-3
Mutations on both RAS and RAF are associated with many cancer types, but the understanding of molecular bases regulating RAS-RAF pathway is still limited.
A crucial step in the activation of RAF kinase is its dephosphorylation at the N terminal, performed by the protein phosphatase PP1C. RAS regulates the process by forming a complex with SHOC2 and PP1C, driving the selectivity towards RAF as substrate of the dephosphorylation.
Genentech team has reproduced in vitro the complex SHOC2-PP1C-RAS and determined its structure through cryo-EM. They found that SHOC2 forms a crescent-shaped architecture and acts like a cradle bringing spatially close PP1C and GTP-bound RAS.
The structural elucidation of the interfaces crucial for the complex formation opens an exciting opportunity for the development of novel drugs.

Research & Image Source: https://www.nature.com/articles/s41586-022-04838-3
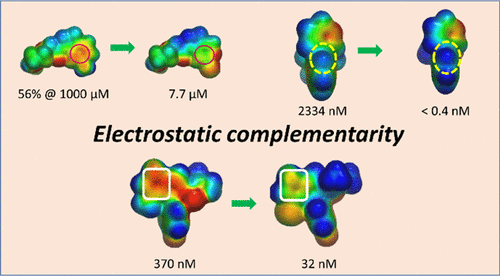
Electrostatic Complementarity in Structure-Based Drug Design
In structure-based drug design, electrostatic complementarity is a (often overlooked) strategy to maximize the binding affinity between small-molecules and their target. In their recent miniperspective (https://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.2c00164), Cons et. al. illustrate some good examples of how this technique can accelerate drug discovery.
Electrostatic potential surfaces (EPS) of the protein binding pocket and the ligand, allow rapid identification of optimizable binding interactions. Charge complementarity-driven design can increase affinity and selectivity, through unusual binding modes. ESP of the ligand in the bounded conformation, can be also exploited to design intramolecular interactions, to stabilize and improve physiochemical properties of the molecule.
This very powerful strategy is sometimes neglected because an accurate calculation ESP is computationally costly and requires high expertise. The recent development of machine/deep learning methods allows a faster generation of ESP data with good quality and less computational effort. This opens unprecedently affordable opportunities for exploiting this powerful technique.
Research & Image Source: https://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.2c00164